3D Molecular Discovery Unleashed: Uni-Mol is Now Available on Vecura
This update enables drug discovery researchers, structural biologists, and computational chemists to generate 3D molecular representations, predict protein-ligand binding poses, and simulate conformations through a guided workflow inside Vecura, without setting up complex technical infrastructure.
What is Uni-Mol?
Uni-Mol is a universal 3D molecular representation learning framework designed to significantly expand the capabilities and application scope of machine learning in drug discovery and quantum chemistry. Unlike traditional methods that treat molecules as 1D sequence strings or 2D topological graphs, Uni-Mol processes 3D atomic coordinates directly using a SE(3)-equivariant Transformer with Gaussian basis kernel functions. It helps users generate highly accurate, geometry-aware molecular representations and predict biological or physical properties. It is especially useful for drug design pipelines, where capturing the spatial, three-dimensional structure of molecules is critical for understanding molecular interactions and docking configurations.
What can users do with Uni-Mol on Vecura?
With Uni-Mol on Vecura, users can:
-
Generate 3D-Aware Molecular Representations: Extract high-quality, fixed-length embedding vectors (such as the 512-dimensional CLS token in Uni-Mol v1 or scalable embeddings from Uni-Mol v2) and per-atom embeddings from SMILES strings for downstream property prediction, molecular similarity analysis, or compound clustering.
-
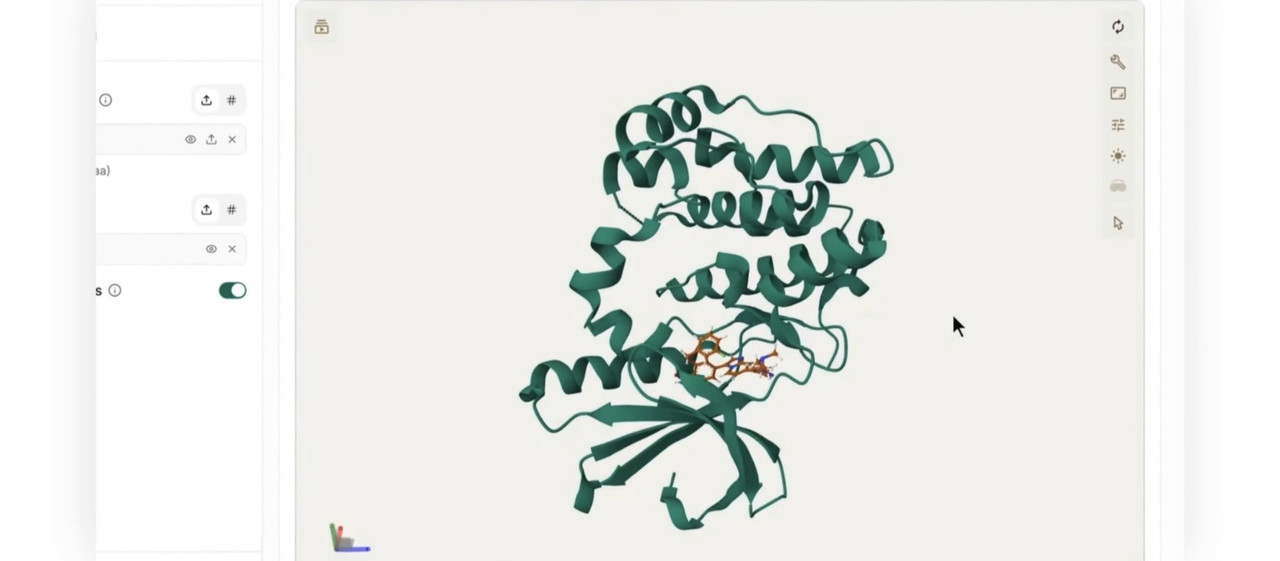
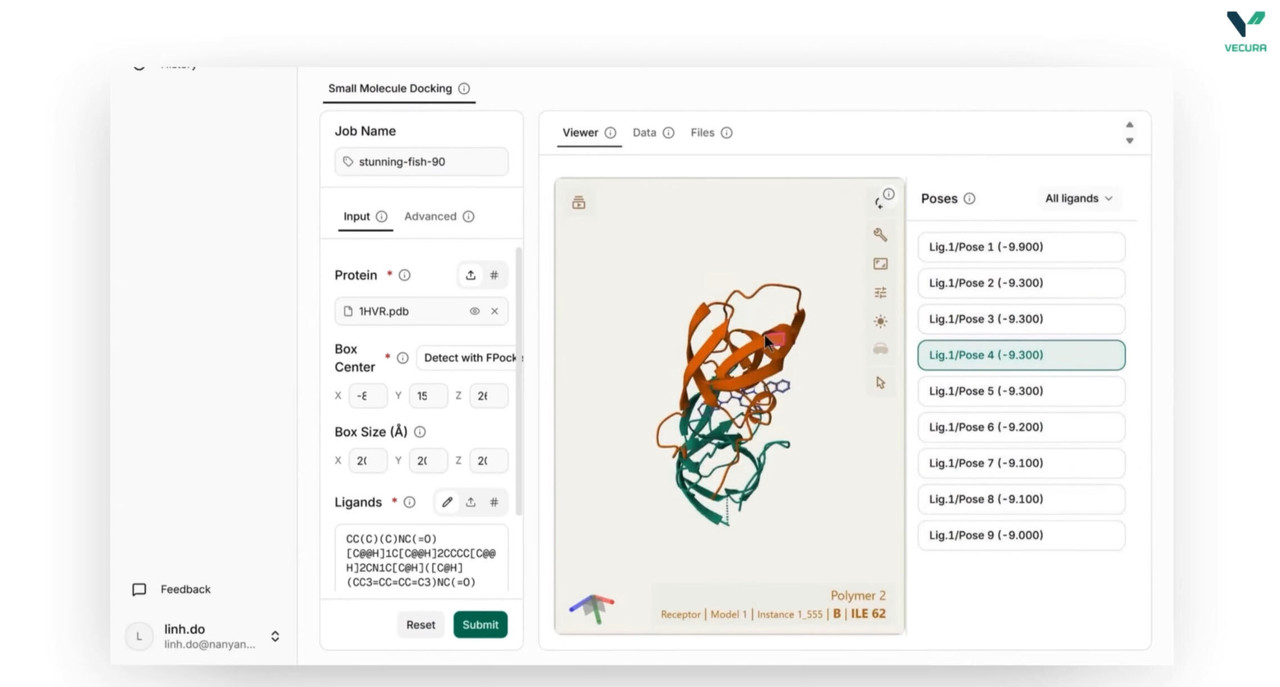
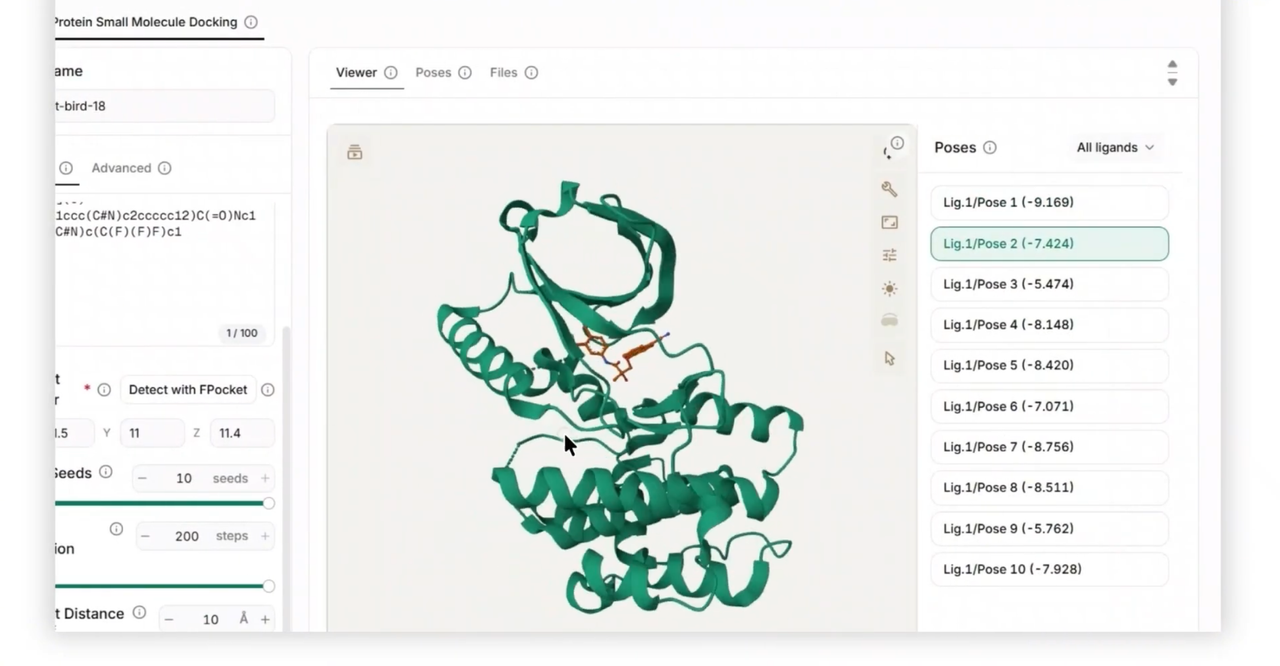
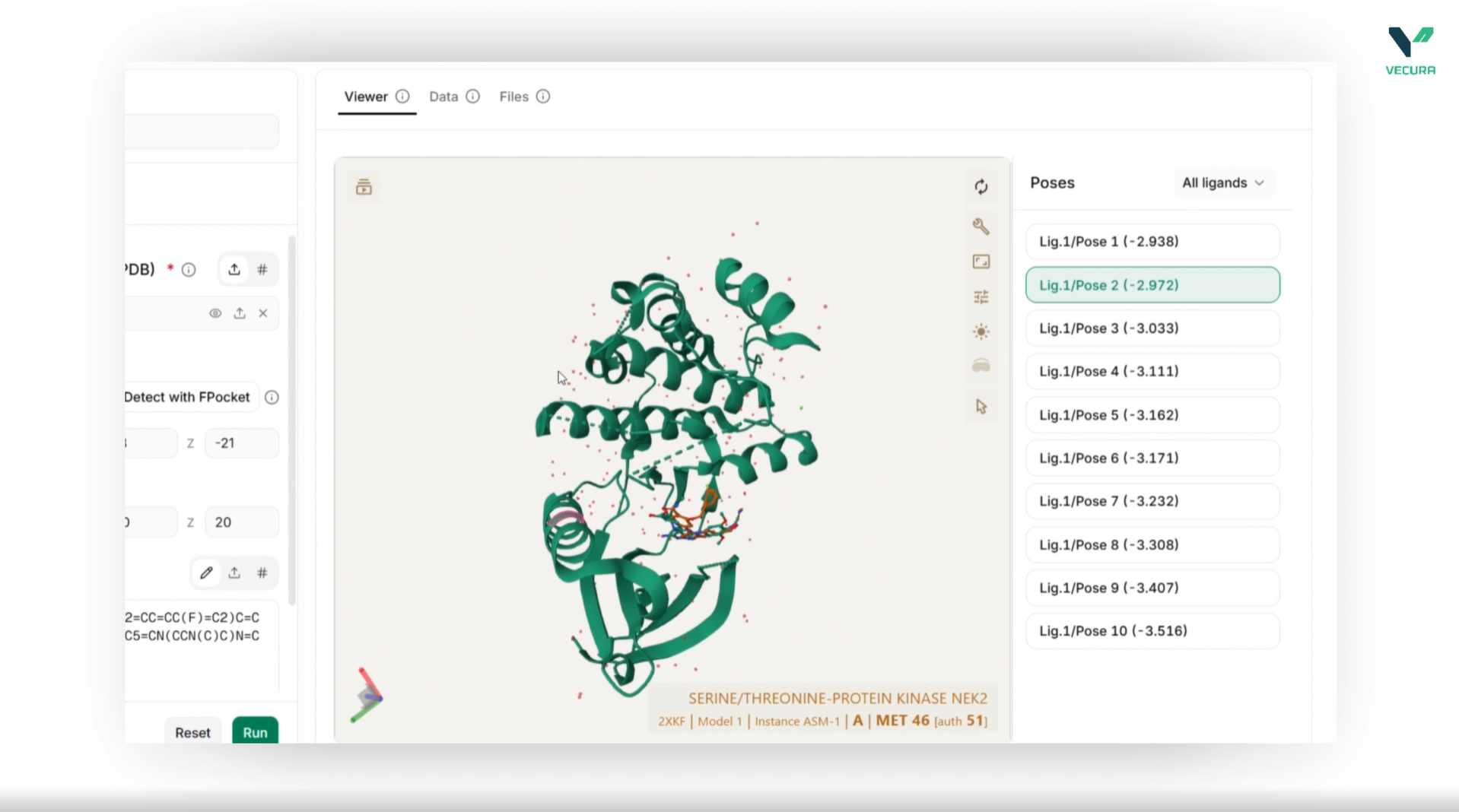
Predict Protein-Ligand Binding Poses: Leverage Uni-Mol Docking V2 to predict how candidate small-molecule ligands dock into a specific protein pocket, identifying favorable binding configurations with remarkable accuracy.
-
Generate Low-Energy 3D Conformations: Generate ranked sets of realistic 3D molecular conformations from standard 2D SMILES strings using models trained on robust chemistry datasets like QM9 and DRUGS.
-
Configure Models and Parameters Seamlessly: Easily toggle between version backbones (Uni-Mol v1 or the scalable Uni-Mol v2 with sizes up to 1.1B parameters), adjust batch sizes, and apply post-docking steric clash relaxation with direct parameter inputs.
What the output means
The output provides 3D molecular structures, ligand binding rankings with confidence scores, high-dimensional representation files, and visualizable spatial coordinates.
This output should be used to support scientific decision making. It does not replace experimental validation.
Why this matters
Traditional virtual screening and molecular modeling heavily depend on simplified 2D graph representations—which ignore crucial spatial and stereochemical realities—or computationally intensive, physics-based simulations that do not scale well. Uni-Mol overcomes these limitations by utilizing deep learning pre-trained on hundreds of millions of 3D atomic structures and protein pockets. This hybrid approach enables high-throughput, geometry-aware molecular analysis at a fraction of the computational cost, achieving state-of-the-art results across numerous property benchmarks, quantum chemistry tasks, and pose prediction competitions.
By making these advanced capabilities accessible directly inside Vecura's guided environment, researchers can bypass the complex tasks of compiling CUDA environments, managing deep learning dependencies, and manually processing molecular geometries. This democratizes state-of-the-art 3D molecular modeling, accelerating candidate screening and structural analysis for scientific teams everywhere.
-
Developed by: DP Technology
-
Source: Official GitHub Repository and Hugging Face Models
-
Reference: Uni-Mol: A Universal 3D Molecular Representation Learning Framework (ICLR 2023), Uni-Mol+ (Nature Communications 2024), and Uni-Mol2 (NeurIPS 2024)
Vecura で Uni-Mol を試す。
モデルワークスペースを開き、ご自身の入力で評価を始めましょう。