AutoDock-Vina Now Available on Vecura for Accelerated Drug Discovery
This update enables computational biologists, medicinal chemists, and drug discovery researchers to predict small molecule binding poses and affinities through a guided workflow inside Vecura, without setting up complex technical infrastructure.
What is AutoDock-Vina?
AutoDock-Vina is an open-source molecular docking program originally designed and implemented by Dr. Oleg Trott in the Molecular Graphics Lab at Scripps Research Institute. It's a widely used tool in computational drug discovery that predicts how small molecule ligands bind to protein targets, estimating both the preferred binding orientation (pose) and binding affinity.
It helps users screen large virtual libraries of drug-like molecules to identify promising candidates for further development. It is especially useful for structure-based drug design, lead optimization, and virtual screening campaigns where rapid evaluation of compound binding is critical.
What can users do with AutoDock-Vina on Vecura?
With AutoDock-Vina on Vecura, users can:
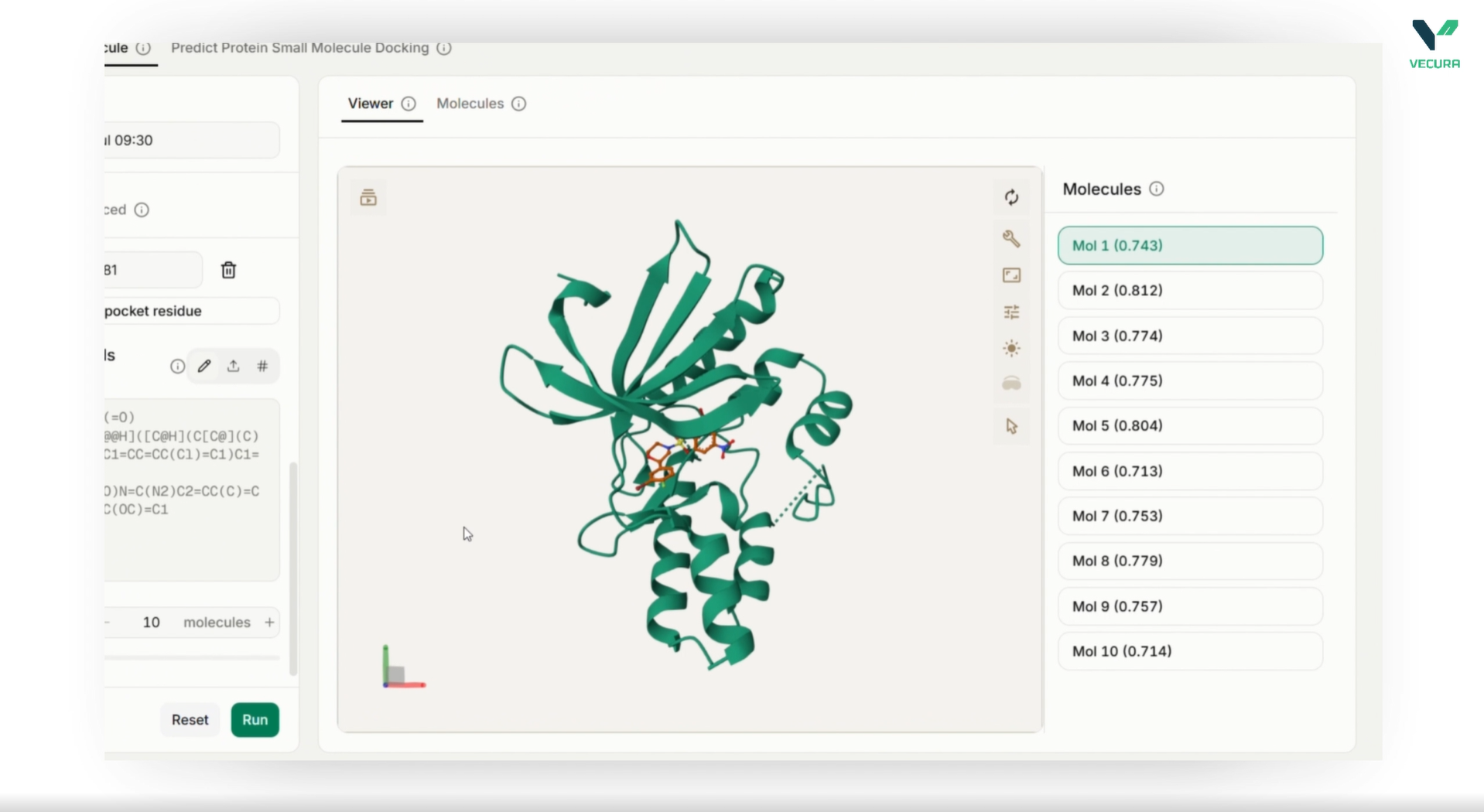
- Predict the preferred binding orientation and affinity of small molecule ligands against target protein receptors
- Perform high-throughput virtual screening of hundreds of ligands in parallel using GPU acceleration
- Define precise docking search boxes around binding pockets using intuitive coordinates and dimensions
- Process ligands from multiple input formats including SMILES strings, CSV files, or RCSB CCD codes
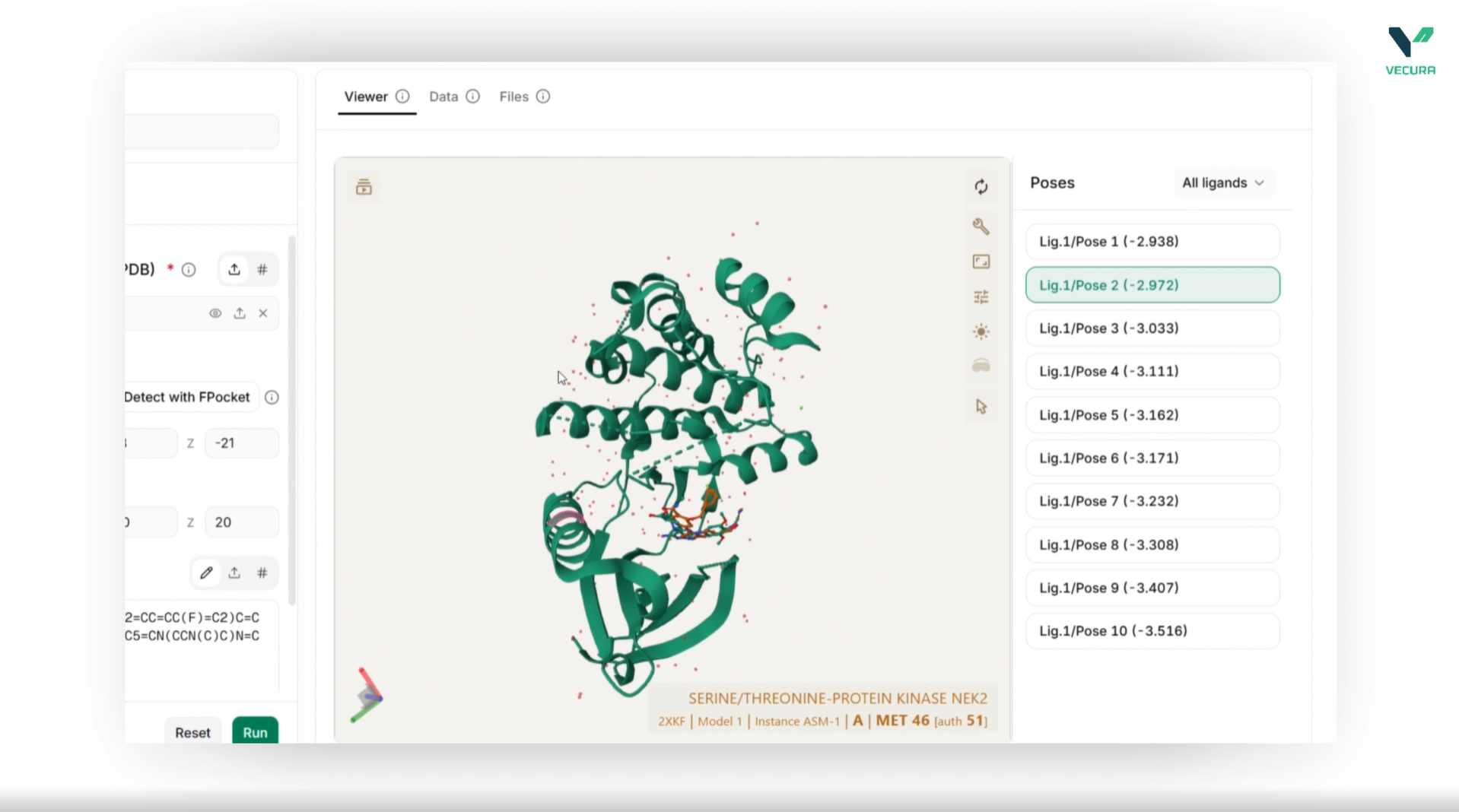
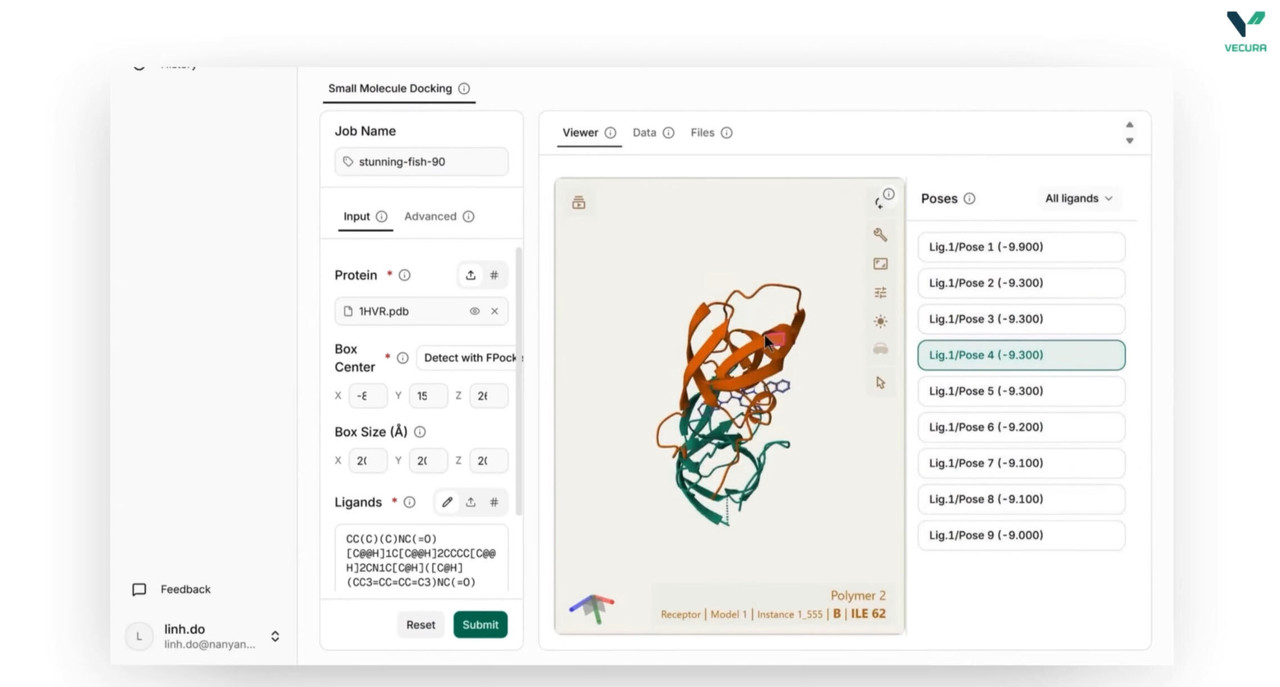
What the output means
The output provides ranked binding poses per ligand with predicted binding affinities in kcal/mol, along with the 3D coordinates of each pose in PDBQT format for visualization and downstream analysis.
This output should be used to support scientific decision making. It does not replace experimental validation.
Why this matters
Molecular docking is a fundamental computational technique in modern drug discovery that accelerates the identification of potential therapeutic candidates. AutoDock-Vina has become a gold standard in the field due to its exceptional balance of speed and accuracy—providing up to two orders of magnitude faster performance than its predecessor while improving binding mode prediction accuracy. Its integration into Vecura democratizes access to this powerful technology, enabling researchers without specialized computational infrastructure to leverage state-of-the-art docking capabilities for structure-based drug design, lead optimization, and virtual screening.
Developed by: Scripps Research Institute (Molecular Graphics Lab) Source: https://vina.scripps.edu Reference: AutoDock Vina: Improving the Speed and Accuracy of Docking (J. Comput. Chem. 2010) https://doi.org/10.1002/jcc.21334
Vecura で AutoDock-Vina を試す。
モデルワークスペースを開き、ご自身の入力で評価を始めましょう。