EquiBind is now available on Vecura
This update enables computational biologists and drug discovery researchers to predict protein-ligand binding structures through a guided workflow inside Vecura, without setting up complex technical infrastructure.
What is EquiBind?
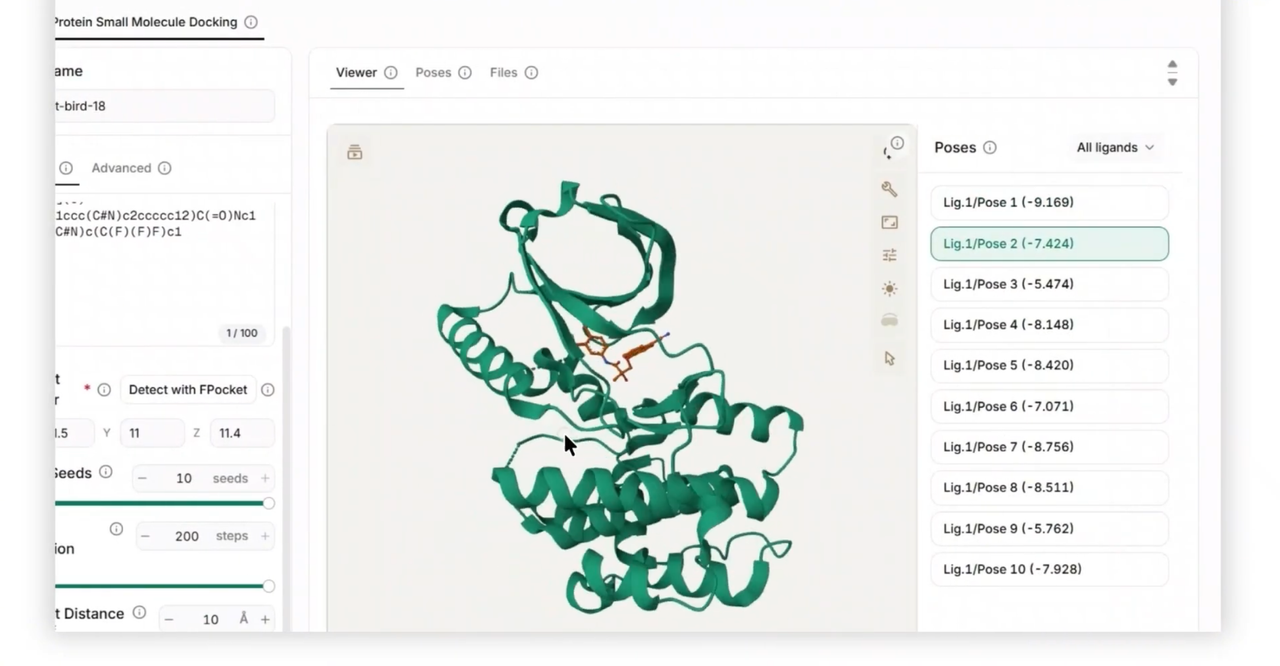
EquiBind is an SE(3)-equivariant geometric deep learning model that performs blind protein-ligand docking. It predicts both the receptor's binding location and the ligand's bound 3D pose and orientation in a single forward pass, without requiring a pre-specified binding pocket. The model operates on 3D point clouds of atoms using graph neural networks that are invariant to global rotations and translations, learning physically meaningful geometry rather than memorizing coordinate frames.
It helps users rapidly predict how small molecules bind to protein targets. It is especially useful for virtual screening campaigns where researchers need to evaluate multiple candidate drug molecules against a receptor structure, achieving millisecond inference times—1,200 times faster than traditional docking tools like AutoDock Vina.
What can users do with EquiBind on Vecura?
With EquiBind on Vecura, users can:
-
Blind-dock small-molecule ligands to receptor proteins without specifying binding pockets
-
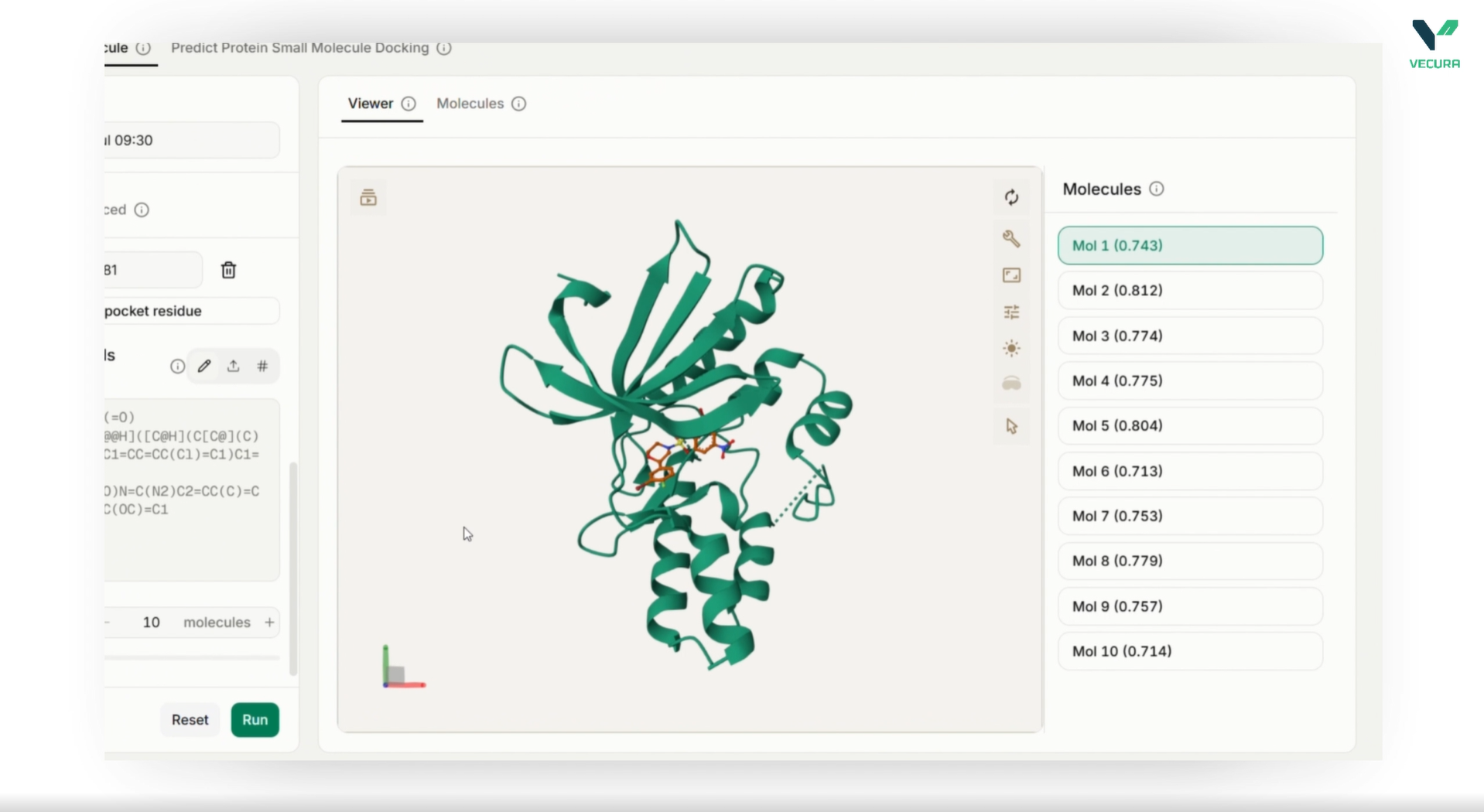
Predict 3D binding poses for single ligands or screen multiple ligands in batch mode
-
Generate docked conformers suitable for visual inspection in molecular viewers (PyMOL, Mol*)
-
Produce starting structures for downstream rescoring workflows (MM-GBSA) or molecular dynamics simulations
What the output means
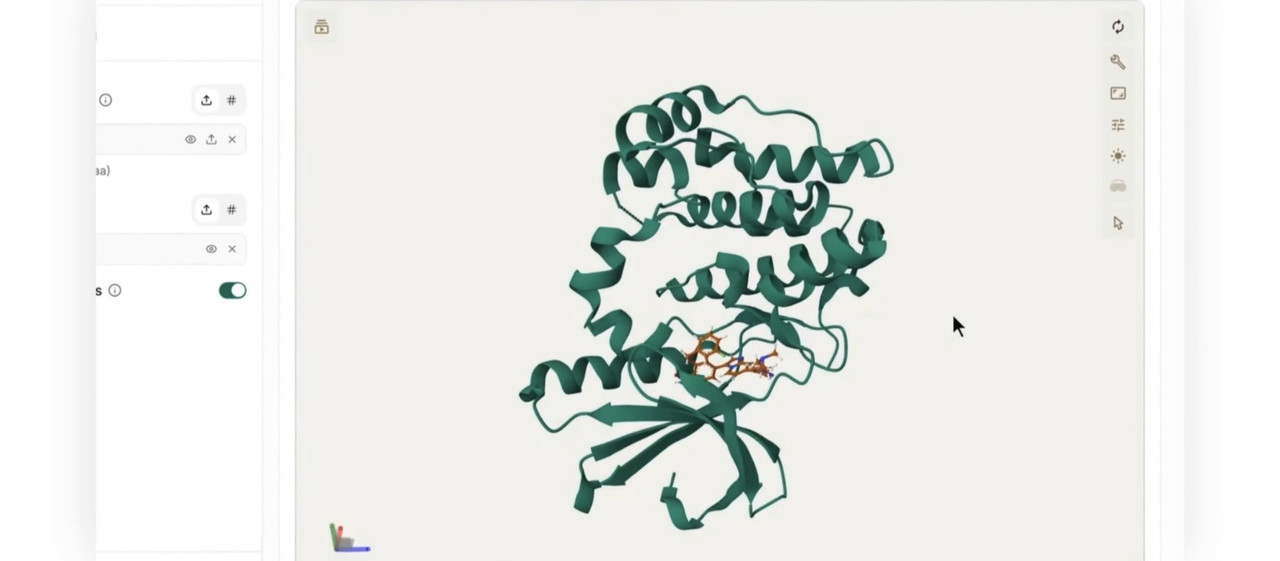
The output provides a docked ligand structure in SDF format, containing the predicted 3D conformation and binding location of the ligand relative to the receptor. For multi-ligand screens, the output includes one docked conformer per successfully processed ligand.
This output should be used to support scientific decision making. It does not replace experimental validation.
Why this matters
Predicting how drug-like molecules bind to protein targets is a fundamental challenge in drug discovery. Traditional docking methods require users to manually specify binding pockets and perform computationally expensive energy minimization searches, taking minutes to hours per complex. EquiBind eliminates these bottlenecks by treating the entire receptor surface as a potential binding site and using geometric deep learning to directly predict the bound structure in milliseconds.
This speed advantage enables researchers to rapidly explore protein-ligand interactions across entire compound libraries, identify novel binding pockets, and accelerate the early stages of drug discovery. While EquiBind provides pose predictions without affinity scoring, the docked structures serve as valuable starting points for more detailed binding affinity calculations and experimental validation studies.
-
Developed by: Hannes Stärk, MIT
-
Source: ICML 2022 paper and GitHub repository
-
Reference: https://arxiv.org/abs/2202.05146 | https://github.com/HannesStark/EquiBind
Try EquiBind on Vecura.
Open the model workspace and start evaluating it with your own inputs.