FAIRChem UMA Now Available on Vecura
This update enables researchers and scientists to predict energy, forces, and stress for atomic systems across a wide range of chemistries through a guided workflow inside Vecura, without setting up complex technical infrastructure.
What is FAIRChem UMA?
FAIRChem UMA (Universal Models for Atoms) is a family of machine-learned interatomic potentials (MLIPs) developed by Meta's FAIR Chemistry. It predicts energy, forces, and stress for atomic systems across a wide range of chemical domains including catalysis, inorganic materials, molecules, metal-organic frameworks (MOFs), and molecular crystals using a single model with task-specific heads. The model is designed to be used through the ASE FAIRChemCalculator interface, making it easy to integrate into existing workflows without requiring complex setup. It helps users perform high-throughput DFT-quality atomistic simulations with reduced computational cost and complexity. It is especially useful for researchers in academia and industry who need to screen large numbers of atomic configurations or run molecular dynamics simulations but are limited by the time and resources required for traditional DFT calculations.
What can users do with FAIRChem UMA on Vecura?
With FAIRChem UMA on Vecura, users can:
-
Predict the energy, forces, and stress for any given atomic configuration from a single molecule to periodic slabs and crystals.
-
Optimize the geometry of an atomic system to a local minimum and retrieve the final relaxed structure.
-
Run NVT or NVE molecular dynamics simulations to explore configurational space and study the dynamic behavior of a system at finite temperature.
-
Access the potential energy surface for various chemistries through a single model, eliminating the need for domain-specific potentials.
-
Integrate seamlessly with ASE (Atomic Simulation Environment) for additional simulation capabilities such as nudged elastic band (NEB) calculations and more.
What the output means
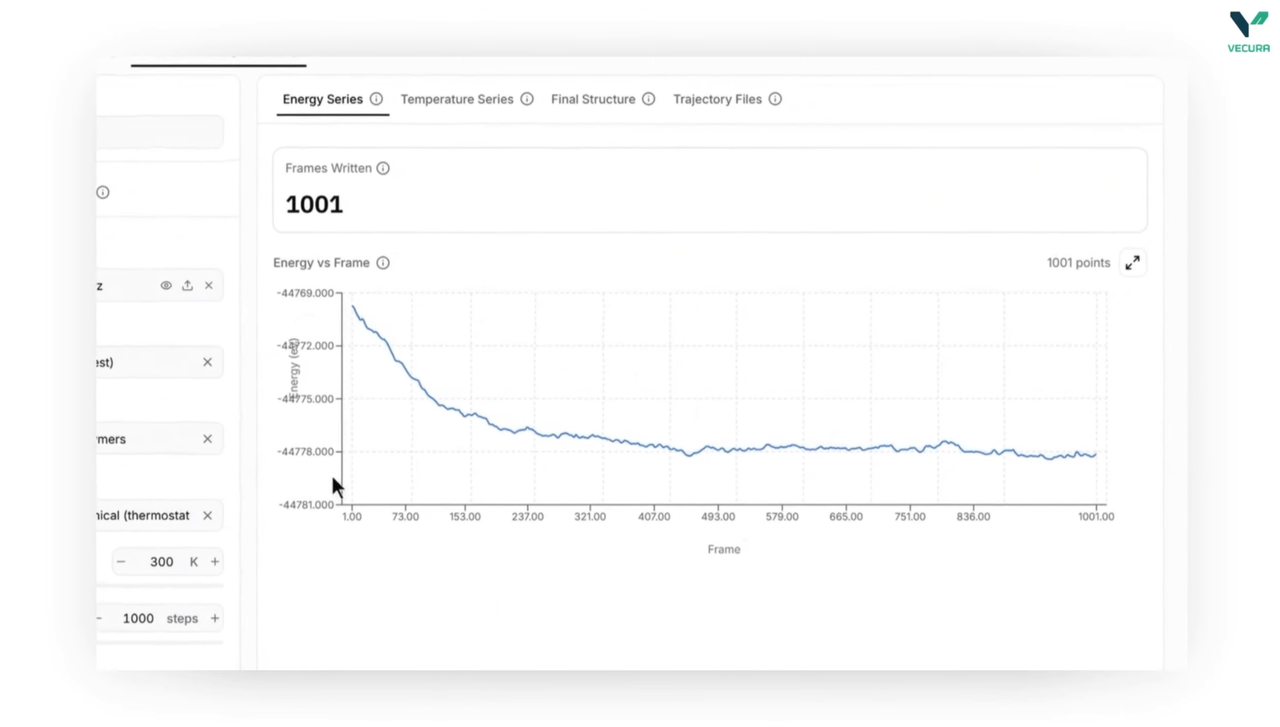
The output provides total potential energy, per-atom forces, and, if applicable, the stress tensor for the given atomic configuration. In the case of geometry optimization, the relaxed atomic structure is also provided. For molecular dynamics, a trajectory file along with inline energy and temperature series is returned. These data points are essential for understanding the stability and reactivity of the system under investigation. This information should be used to support scientific decision-making; however, it does not replace the need for experimental validation.
Why this matters
The development of universal models like FAIRChem UMA is significant because it enables scientists to accelerate their research by providing fast, accurate, and versatile predictions for a broad range of atomic systems. This can lead to faster discovery cycles in materials science, catalysis, and other areas where understanding atomic-scale phenomena is crucial. By reducing the reliance on computationally expensive DFT calculations, researchers can focus more on the creative aspects of their work, such as designing new materials or optimizing reaction pathways, while still maintaining a high level of accuracy in their predictions.
-
Developed by: Meta's FAIR Chemistry
Try FAIRChem UMA on Vecura.
Open the model workspace and start evaluating it with your own inputs.