LigandMPNN is Now Available on Vecura
This update enables structural biologists, therapeutic designers, and researchers to design custom protein sequences conditioned on bound ligands, metals, and nucleic acids through a guided workflow inside Vecura, without setting up complex technical infrastructure.
What is LigandMPNN?
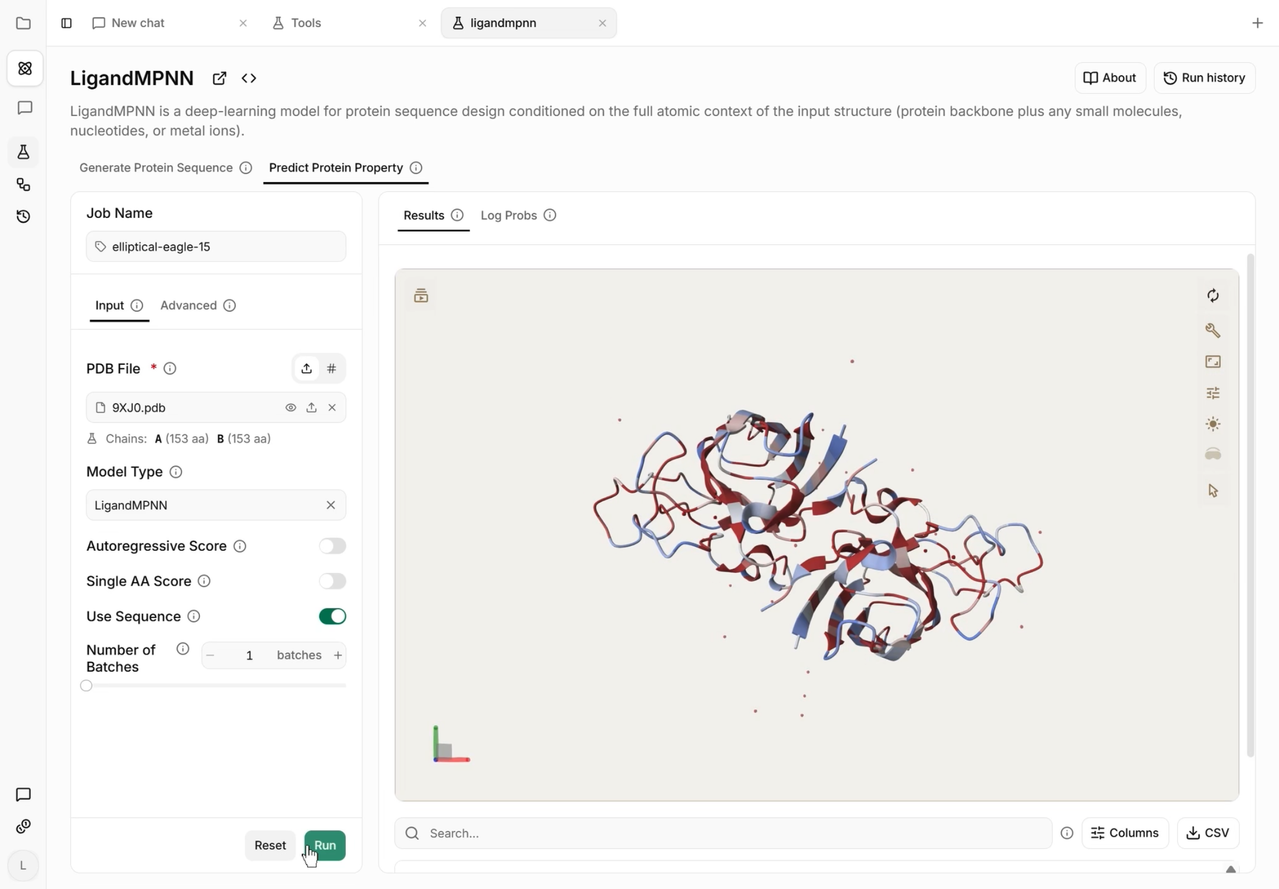
LigandMPNN is a deep-learning model designed to solve the protein sequence design problem by explicitly conditioning on the complete atomic context of an input structure. Unlike its predecessor, ProteinMPNN, which models sequence design solely based on the protein backbone, LigandMPNN represents non-protein elements—such as small-molecule ligands, cofactors, metal ions, and nucleotides—as a point cloud of atoms appended directly to the protein graph. It helps users generate optimized amino acid sequences that maintain high chemical and structural complementarity to these crucial target molecules. It is especially useful for designing novel enzymes, metal-binding sites, biosensors, and therapeutic interfaces where interactions with non-protein entities are key.
What can users do with LigandMPNN on Vecura?
With LigandMPNN on Vecura, users can:
-
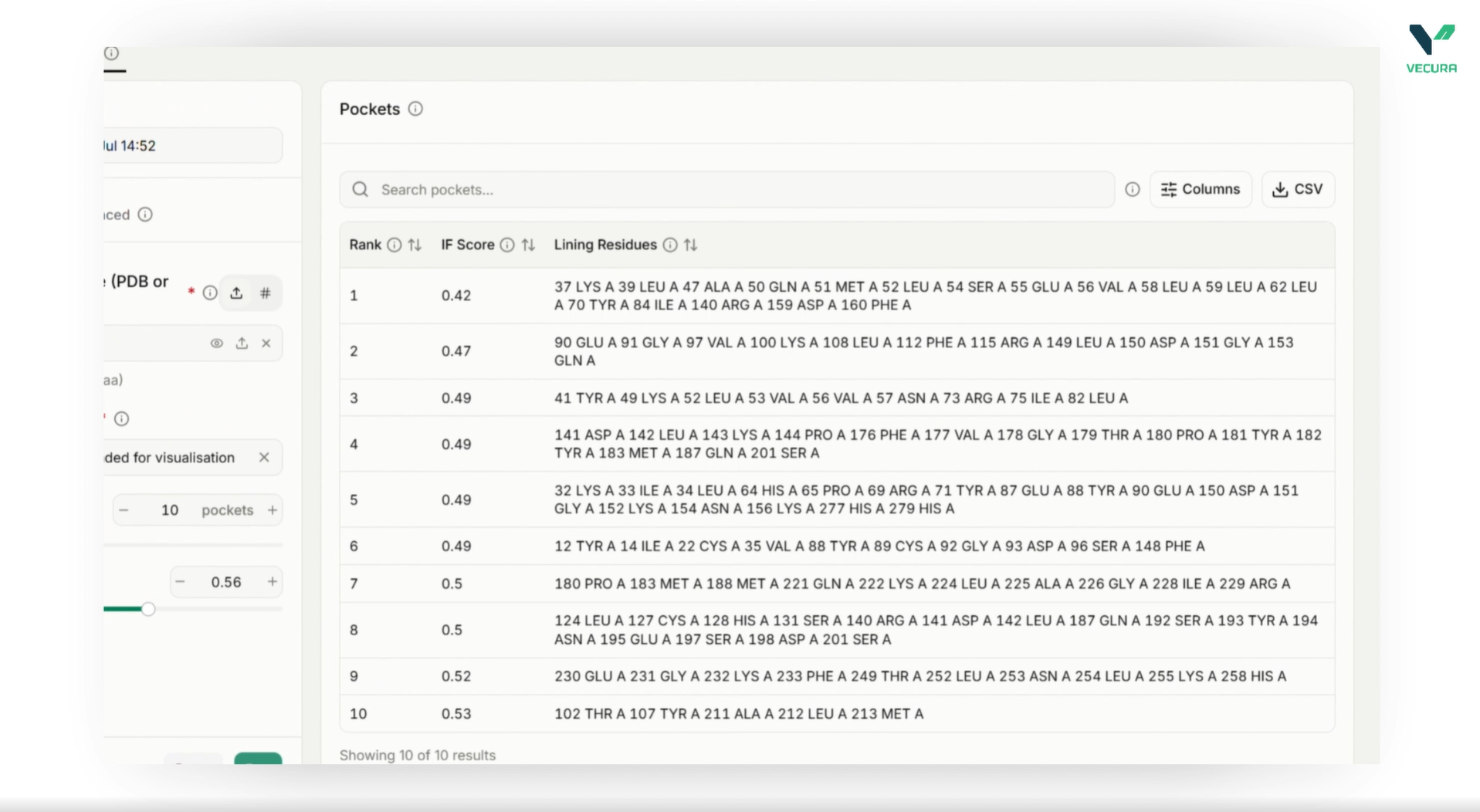
Design sequences conditioned on full atomic environments: Generate sequences tailored to the exact coordinates of small molecules, cofactors, metal ions, and DNA/RNA partners.
-
Target and customize specific residues: Pinpoint which chains or specific residue positions should be redesigned while keeping the rest of the protein sequence fixed.
-
Incorporate design constraints and biases: Apply custom global amino acid biases, exclude specific residues (like cysteines) from the final output, and enforce homomeric or heteromeric symmetry.
-
Refine side chains and score sequences: Run integrated side-chain packing modules to output ready-to-inspect 3D structures or evaluate existing sequences by calculating per-residue log-likelihoods.
What the output means
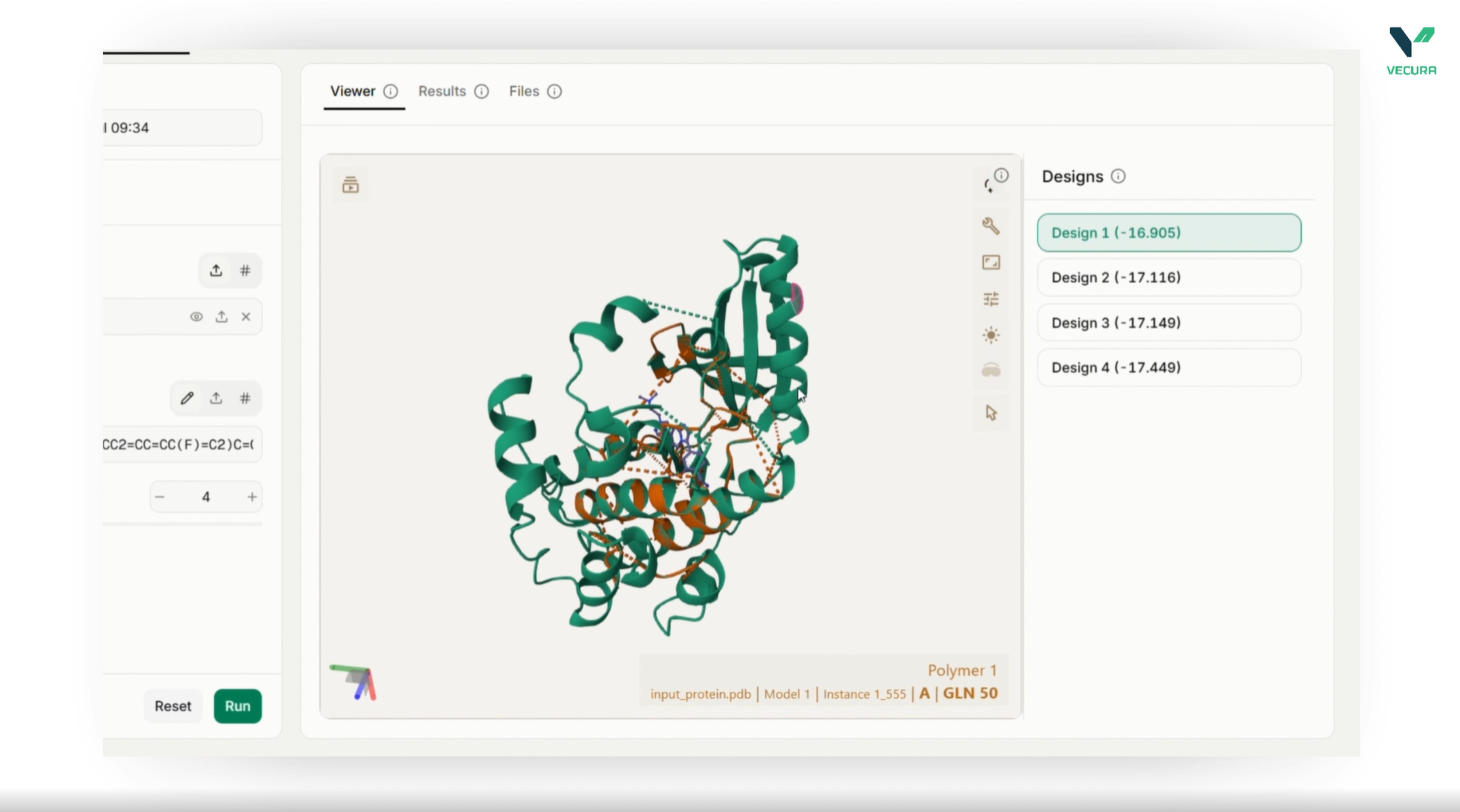
The output provides generated, designed amino acid sequences along with confidence scores and 3D structural files. Specifically, users receive the designed_sequences accompanied by overall confidence values (as well as ligand-specific confidence metrics for residues near the target molecule), a multi-record FASTA file containing all designs, and a representative designed_pdb structure file. When run in side-chain packing mode, this structure contains full-atom coordinates including optimized side-chain placements, allowing immediate visual inspection. Additionally, scoring mode outputs per-residue log-probability matrices (log_probs and probs) representing the likelihood of each amino acid at each sequence position.
This output should be used to support scientific decision making. It does not replace experimental validation.
Why this matters
In traditional protein design, computational models often ignore the presence of non-protein atoms, treating ligands, nucleic acids, and metal ions as simple "empty space" or requiring cumbersome, physics-based hand-parameterization for every new ligand. This omission has historically limited the reliability of designed binding pockets, active sites, and interface regions. LigandMPNN overcomes these bottlenecks by treating ligand atoms as an unrestricted point cloud appended to the graph, meaning it requires no pre-defined covalent bond topologies or chemical rules. This lets it generalize effortlessly to entirely novel chemical classes and complex multi-state systems.
By bringing LigandMPNN to the Vecura platform, we remove the technical overhead of configuring GPUs, resolving CUDA and Python dependencies, and scripting inputs. Biologists and drug designers can now seamlessly navigate structural sequence design tasks, accelerating the transition from digital protein generation to wet-lab screening of optimized binders and biocatalysts.
-
Developed by: Justas Dauparas and the David Baker Lab at the University of Washington (Institute for Protein Design)
-
Source: Official GitHub Repository
在 Vecura 上试用 LigandMPNN
打开模型工作区,用您自己的输入开始评估